Pervaiz Akhtar Shah1,
Sajid Bashir2,
Muhammad Ahsan1 ![]() ,
Nasir Abbas1,
Muhammad Zubair Malik2,
Hafiz Muhammad Irfan Nazar1
,
Nasir Abbas1,
Muhammad Zubair Malik2,
Hafiz Muhammad Irfan Nazar1
For correspondence:- Muhammad Ahsan Email: ahsanshareef105@hotmail.com Tel:+923334060054
Received: 19 November 2015 Accepted: 20 March 2016 Published: 30 April 2016
Citation: Shah PA, Bashir S, Ahsan M, Abbas N, Malik MZ, Nazar HM. Bioequivalence evaluation of new microparticulate capsule and marketed tablet dosage forms of lornoxicam in healthy volunteers. Trop J Pharm Res 2016; 15(4):877-883 doi: 10.4314/tjpr.v15i4.30
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose:To compare oral bioavailability and pharmacokinetic parameters of different lornoxicam formulations and to assess similarity in plasma level profiles by statistical techniques.
Methods:An open-label, two-period crossover trial was followed in 24 healthy Pakistani volunteers (22 males, 2 females). Each participant received a single dose of lornoxicam controlled release (CR) microparticles and two doses (morning and evening) of conventional lornoxicam immediate release (IR) tablet formulation. The microparticles were prepared by spray drying method. The formulations were administered again in an alternate manner after a washout period of one week. Pharmacokinetic parameters were determined by Kinetica 4.0 software using plasma concentration-time data. Moreover, data were statistically analyzed at 90 % confidence interval (CI) and Schuirmann’s two one-sided t-test procedure.
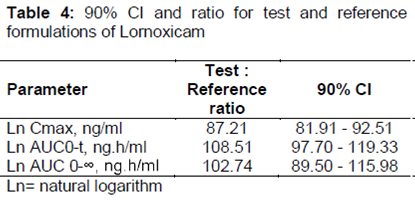
Results:Peak plasma concentration (Cmax) was 20.2 % lower for CR formulation compared to IR formulation (270.90 ng/ml vs 339.44 ng/ml, respectively) while time taken to attain Cmax (tmax) was 5.25 and 2.08 h, respectively. Area under the plasma drug level versus time (AUC) curve was comparable for both CR and IR formulations. The 90 % confidence interval (CI) values computed for Cmax, AUC0-24, and AUC0-¥, after log transformation, were 87.21, 108.51 and 102.74 %, respectively, and were within pre-defined bioequivalence range (80 - 125 %).
Conclusion:The findings suggest that CR formulation of lornoxicam did not change the overall pharmacokinetic properties of lornoxicam in terms of extent and rate of lornoxicam absorption.
Introduction
Lornoxicam is a short-acting non-steroidal anti-inflammatory drug (NSAID) which belongs to the oxicam group. It decreases prostaglandin production by reducing cyclo-oxygenase (COX) activity. It is successfully being used in clinical settings for alleviating the symptoms of rheumatoid arthritis, osteoarthritis, acute sciatica and ankylosing spondylitis. Moreover, it has demonstrated comparable analgesic efficacy to morphine for relieving postoperative pain [3]. Due to short half-life of 3 to 5 h, the drug is highly feasible to be formulated as controlled release formulation [1,2].
Patient compliance to prescribed regimen of NSAIDs in chronic disorders (rheumatoid arthritis & ankylosing spondylitis) is generally poor [4]. The reasons for low compliance of NSAIDs include temporary relief of symptoms and gastric irritation associated with frequent intake of NSAIDs. Substantial variation in compliance has been observed in patients taking NSAIDs resulting in poor drug efficacy and safety [5].
Previous studies have revealed that switching from IR to CR formulations reduces the side effects as well as improves outcomes of therapy [6-8]. However, no report demonstrates in-vivo comparative pharmacokinetic analysis for any NSAID drug by formulating different release formulations. Therefore, an attempt was made to compare pharmacokinetic parameters (tmax, Cmax, AUC) of newly formulated CR lornoxicam microparticles with IR tablet counterpart.
Methods
Preparation of lornoxicam-loaded microparticles
Lornoxicam microencapsulation was achieved by spray drying according to the polymer blend concentrations specified in table 1. We dissolved weighed quantities of polymers in acetone and ethanol mixture used in the ratio of 40:60. Hydroxy propyl methyl cellulose (HPMC) 15 cps was dissolved first in organic mixture followed by Eudragit L100 and clear solution was obtained. Then drug was added into polymer solution with continuous stirring for 45 - 60 min. Finally, drug polymer dispersion so prepared was spray-dried using Lab scale spray dryer with a nozzle of 2 mm diameter (YC 1500, Shangai, China). The dried particles enclosing core material were collected in the bottom of dryer [9]. The parameters set for spray drying were as follows: Inlet air temperature, 80 – 90 oC; inlet air volume, 200 - 300m3/h; spray rate, 8-10gm/min; product temperature, 40 - 50 oC; and atomizing air pressure; 1 bar.
Characterization of microparticles
The formulated microcapsules were characterized for morphology, encapsulation efficiency, percent drug loading and in-vitro dissolution studies as described by Shah et al [9].
Subjects’ inclusion and exclusion criteria
Twenty four healthy subjects {age, 21.69 ± 2.32 years (range, 17.18 - 26 years); weight, 70.14 ± 7.98 kg (range, 60.12 - 89 kg); height, 180.25 ± 7.32 cm (range, 168.2 - 190.93 cm); and BMI, 21.59 ± 2.04 kg/m2 (range, 18.73 - 24.82 kg/m2)} were selected for the study. All the subjects were examined to determine their health status based on medical history, physical examination, vital signs, and laboratory tests (hematology, biochemistry, hepatic function, and urinalysis). Subjects who had a history of gastric ulcers and those receiving oral corticosteroids were not included in the study. Written informed consent was taken from each participant before the commencement of study. They were free to withdraw from the study at any time. Physical examination took into account normal laboratory tests conducted before, during and after study.
Study design
The present open label, randomized, two-way crossover, comparative bioavailability study was performed according to the principles of Helsinki Declaration (WMA) [10] and Good Clinical Practices (GCP) [11]. The study protocols were approved by the ethical committee of Human Research Review Board of University College of Pharmacy, University of the Punjab, Lahore, and the guidelines of Helsinki Declaration were followed. All the volunteers were fasted overnight at least 10 h prior to dosing with water as desired. Subjects received single dose of CR formulation and two doses of IR formulation followed by a washout period of 7 days and administration of alternate formulations. Lornoxicam IR formulation was administered twice after interval of 12 h in each period. None of the participants was on medication of any sort which may interfere with the release and detection of drugs under study. Standard meal was provided after 4 h of drug administration.
Blood sampling and processing
At predetermined time interval, 5ml of blood was withdrawn at 0, 0.5, 1, 2, 3, 5, 8, 10, 12, 13, 14, 15, 18, 20 and 24 h by phlebotomists. Disposable 5 ml syringes were used to collect blood samples into heparin containing centrifuge tubes. The samples, immediate after collection, were centrifuged at 4000 rpm for 10 min to separate plasma which was carefully conveyed to eppendrof tubes using micropipette. The prepared plasma samples were kept at −20 oC until lornoxicam concentration was estimated by bio-analytical HPLC method.
Determination of drug in blood samples
Lornoxicam levels in plasma samples were determined by specific and sensitive HPLC method. The mobile phase used was mixture of phosphate buffer (pH 4.5, adjusted with orthophosphoric acid) and acetonitrile (45:55, v/v). Freshly prepared mobile phase was degassed by vacuum filtration, sonicated and pumped with a flow rate of 1 ml/min. After adding 0.5 ml of methanol, 0.5 ml of trichloroacetic acid and 0.5 ml of internal standard solution (piroxicam, 20 µg/ml), plasma samples were vortex mixed for 3 min and then centrifuged at 2500 rpm for 5min. Then, samples were placed on extrelut-1 columns and dichloromethane (10 ml) was used to elute lornoxicam. The eluate was dehydrated under nitrogen stream (35 oC) followed by reconstitution with 400 μl of mobile phase. Finally, after centrifugation at 100 rpm, 50 μl supernatant was injected into chromatographic system.
HPLC system consisted of hypersil ODS column (150 × 4.5 i.d, 5µm particle size, Agilent) for carrying out separation. Class GC software was connected with variable wavelength UV detector for data processing and chromatographic integration. An isocratic pump (LC-10 ATVP, Shimadzu Corporation, Japan) was used for passing the mobile phase through the column.
Tolerability assessment
Physical assessment and vital signs (pulse rate, blood pressure and body temperature) were monitored for each enrolled subject before administration of study drugs and during the blood sampling. Subjects were continuously examined by the physicians throughout the study for incidence of adverse effects.
Pharmacokinetic and statistical analyses
The parameters included Cmax, AUC0-24, AUC0-¥, tmax, half-life (t1/2), mean residence time (MRT), and elimination rate constant (ke) were determined by Kinetica 4.0 (Thermofisher Scientific, PK/PD software, USA) for each subject. Tmax and Cmax were also estimated directly from the constructed plot of plasma concentration- time profiles [12,13].
Significant difference in parameters was considered at p < 0.05. Analysis of variance (ANOVA) for a crossover (2 × 2) study design was calculated by SPSS software (IBM Statistics 21, USA). The formulations were considered comparable if 90 % CI for the geometric mean ratios (test: reference) of ln-transformed Cmax, AUC0-t, and AUC0-¥ rest between 0.80 and 1.25 - a stipulated criterion of US Food and Drug administration [14,15]. Average bioequivalence was further confirmed between test and reference formulations by applying Schuirmann two 1-sided tests [16].
Results
Characteristics of microparticles
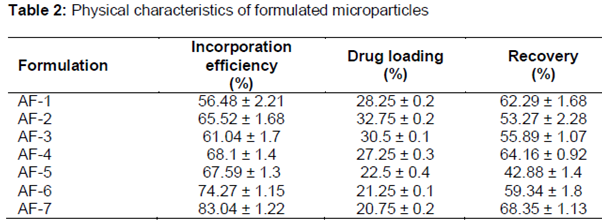
The microparticles were ellipsoidal in shape with surface morphology clearly showing presence of drug particles (). The incorporation efficiency increased from 56.48 % ± 2.21 to 83.04 % ± 1.22 with the increase in the polymer contents. Percent loading was in the range of 42.88 % ± 1.4 to 68.35 % ± 1.13 and was independent of contents of polymer blend.
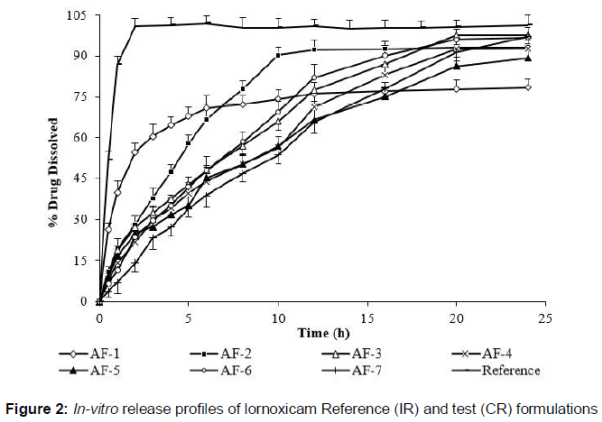
The release results showed that burst release was reduced in microparticles containing higher contents of polymers. This was due to decrease in surface associated drug. Microparticle formulations containing drug polymer blend in the ratio of 1:1 and 1:1.5 released 97.55 ± 3.33 % and 92.6 ± 3.92 % of drug, respectively at the end of 20 h without subsequent release. Similarly, formulation AF-6 liberated maximum drug amount of 96.16 ± 3.88 % on the time point of 20 h. Only formulation AF-7 (1:3) sharply demonstrated drug release of 97.22 ± 3.22 % after 24 h that was a typical sustained release pattern. Taken together, the release pattern clearly showed that rate of drug release but not the extent was decreased on increasing amounts of polymer matrices. Out of seven formulations, microcapsules of one formulation (AF-7) were selected for in-vivo study based on promising results in terms of encapsulation efficiency and release studies. Moreover, the optimized formulation demonstrated near zero order kinetics.
Finally, microparticles (AF-7) equivalent to 16 mg of lornoxicam were filled in 0 size capsules which acted as test formulation. Whilst, reference formulation was immediate release film coated tablets of lornoxicam (8 mg). The in-vitro release profiles of test and reference formulations are shown in .
Validation parameters of developed HPLC method
Intra- and inter-day precision and recoveries (absolute and relative) of lornoxicam in human plasma are shown in .
Pharmacokinetic properties
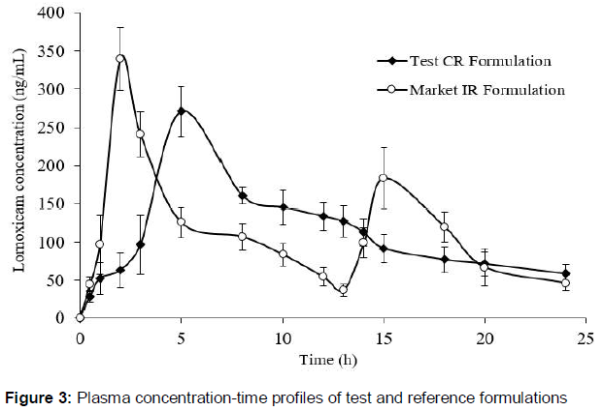
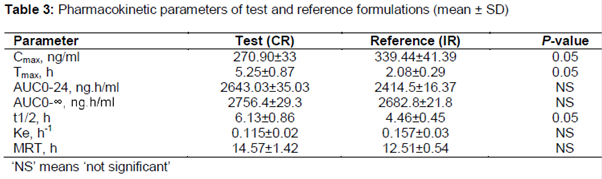
The plasma profiles (mean ± SD) of test and reference formulations of lornoxicam were presented in . The main pharmacokinetic parameters calculated were summarized in table 3. The mean (± SD) Cmax values for test and reference formulations were 270.90 ± 33 ng/mL and 339.44 ± 41.39 ng/mL (p < 0.05), respectively. The mean AUC0–t values in the test and reference formulations were 2643.03 ± 35.03 and 2456.62 ± 25.39 ng.h/mL, and the mean AUC0-¥ were 2740.89 ± 33.03 and 2640.98 ± 27.91 ng.h/mL. The median (range) tmax was 5.3 (5.20±8.30) and 1.93 (1.60–2.30) hours (p < 0.05). The ke was 0.115 (0.02) and 0.157 (0.02) h–1, and the t1/2 was 6.13 (0.86) and 4.46 (0.45) hours (p < 0.05). ANOVA shows no effect of sequence or period on the pharmacokinetic parameters. Surprisingly, the Cmax for first dose of lornoxicam IR formulation was higher than that of second dose. This observation might relate to the influence of circadian rhythms on the kinetics of drugs leading to faster absorption in the morning compared with the evening [17].
Comparative pharmacokinetic profile
The analysis of 90 %CI of Cmax, AUC0-t, and AUC0-¥ values for the test and reference formulations were 81.21 to 92.51 %, 97.70 to 119.33 %, and 89.50 to 115.98 %, respectively and were listed in . The values were well within acceptable bioequivalence criteria (80 - 125 %). The relative bioavailability of lornoxicam was 107.62 %. The findings of Schuirmann test also supported bioequivalence as shown in .
Tolerability
Mild gastrointestinal problem (nausea) occurred in couple of volunteers receiving IR tablet during the second period of the study. Literature survey revealed that this mild side effect was associated with Lornoxicam salt (chlortenoxicam). The problem was immediately overcome by appropriate action of clinician and the subject successfully completed the study. CR formulation was found to lessen the side effects (nausea, diarrhea) of the salt as no active complaint was noticed from participants taking CR formulations. No other treatment related adverse event was recorded throughout the study.
Discussion
This study demonstrated the bioequivalence of two oral formulations of lornoxicam - a newly developed CR microcapsule formulation and a marketed IR tablet - in healthy Pakistani volunteers. AUC0-t and AUC0-¥ are indicators of total amount of absorption, Cmax reflects both extent and rate of absorption, and tmax shows rate of absorption and elimination [18,19]. The Cmax and tmax of the marketed lornoxicam tablet (reference) were comparable with previous reports [20,21]. The comparable Cmax, AUC0–24 and AUC0–¥ for lornoxicam CR and IR formulations showed that both formulations had similar apparent oral clearance. As expected, it was observed that transforming the dosage forms did not change total absorption of lornoxicam even with dose administered once. Moreover, a significant drop of 20.2 % in the mean value of Cmax was observed for lornoxicam CR formulation than for lornoxicam IR (270.90 ± 33 ng/mL and 339.44 ± 41 ng/mL, respectively). This pattern clearly indicates controlled release of lornoxicam from microparticles. Furthermore, these findings proposed that, at steady state, lornoxicam CR and IR formulations will demonstrate comparable bioavailability when administered at the same daily dose (16 mg/day). A considerable difference was seen in the values of t1/2 and ke between the test and reference formulations, further corroborating the profound effect on parameters due to modification in the formulation.
The time to reach Cmax (tmax) was 5.30 h for lornoxicam CR compared with 2.0 h for lornoxicam IR. In future, such delay in action can be avoided by incorporation of small IR fraction in microparticle dosage form. The longer elimination half-life of single CR dose compared with twice-dose of IR formulation (6.13 ± 0.86 h vs 4.46 ± 0.45 h, respectively) suggested that controlled release formulation is suitable for once daily dosing.
The geometric mean of lornoxicam CR/lornoxicam IR ratio for Cmax, AUC0–t and AUC0-¥ were 87.21, 108.51 and 102.74 (90 % CIs 81.91 ± 92.51 %, 97.7 ± 119.3 % and 89.5 ± 115.98 % respectively), which were within bioequivalence pre-defined range (80 %–125 %) with respect to total amount of lornoxicam.
The study has some limitations as the pharmacokinetic data were obtained from healthy Pakistani volunteers so the findings of the study cannot be generalized to all patient populations. Furthermore, serum concentrations of 5-hydroxylornoxicam were not measured due to lack of clinical significance. Small sample size and open-label design were also study limitations.
Conclusion
The findings of this non-replicated crossover study demonstrates that test lornoxicam microparticulate (CR capsules, OD) and reference (IR tablet, BD) formulations at the same molar dose of 16 mg possess comparable pharmacokinetic properties. The good controlled release properties of the microparticle formulation produced by spray drying recommend this approach for the design of once-daily administration of the drug to achieve prolonged analgesia in chronic painful disorders.
Declarations
Acknowledgement
References
Archives


News Updates