Zhaobing Liu,
Junli Huang,
Qing Ding,
Yan Yang,
Hanxiao Sun ![]()
For correspondence:- Hanxiao Sun Email: sunhx718@126.com Tel:+862038375022
Received: 1 February 2016 Accepted: 21 May 2016 Published: 28 June 2016
Citation: Liu Z, Huang J, Ding Q, Yang Y, Sun H. Analysis of contributions of herpes simplex virus type 1 UL43 protein to induction of cell-cell fusion. Trop J Pharm Res 2016; 15(6):1137-1144 doi: 10.4314/tjpr.v15i6.4
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To investigate whether UL43 protein, which is highly conserved in alpha- and gamma herpes viruses, and a non-glycosylated transmembrane protein, is involved in virus entry and virus-induced cell fusion.
Methods: Mutagenesis was accomplished by a markerless two-step Red recombination mutagenesis system implemented on the Herpes simplex virus 1 (HSV-1) bacterial artificial chromosome (BAC). Growth properties of HSV-1 UL43 mutants were analyzed using plaque morphology and one-step growth kinetics. SDS-PAGE and Western blot was employed to assay the synthesis of the viral glycoproteins. Virus-penetration was assayed to determine if UL43 protein is required for efficient virus entry.
Results: Lack of UL43 ex
Conclusion: Thus, these results suggest an important role for UL43 protein in mediating virus-induced membrane fusion and efficient entry of virion into target cells.
Introduction
Herpes simplex virus type 1 (HSV-1) is a common human pathogen that causes most forms of non-genital herpes simplex infection. HSV-1 facilitates virus entry and cell-to-cell spread by mediating fusion which require the intervention of three viral fusogenic glycoproteins, gB, gH, and gL [1,2], and occurs downstream of the interaction of gD with one of the entry receptors [3]. Most mutations that cause extensive virus-induced cell fusion (syncytial or syn mutations) have been mapped to the four HSV genes, UL20 [4], UL24 [5], UL27 encoding glycoprotein B (gB) [6], and UL53 coding for glycoprotein K (gK) [7]. Little is known of the molecular mechanisms by which HSV regulates its own fusion activity.
UL43 gene of HSV-1 encodes a non-glycosylated transmembrane protein which is conserved only in alpha- and gamma herpes viruses [8]. The HSV-1 UL43 has been shown to be non-essential for virus growth in cell culture and deletion of UL43 did not impair viral replication in a mouse infection model [9]. It has been reported that Pseudorabies virus (PrV) UL43 protein strongly inhibited membrane fusion induced by the viral fusion machinery in a transient expression-fusion system [10]. Although the transient co-expression assay does not accurately model viral fusion, it is conceivable that UL43 protein may be involved in modulating membrane fusion.
In this work, we utilized mutant viruses to assess whether the deletion of UL43 gene of HSV-1 affected the ability of dominant syncytial mutations in either gB or gK to cause extensive virus-induced cell fusion. We showed that in viral infections UL43 protein is required for virus-induced cell fusion. Moreover, mutant viruses lacking UL43 gene exhibited slower kinetics of entry into Vero cells than the HSV-1(F) BAC, suggesting that UL43 protein functions in membrane fusion phenomena during both virus-induced cell fusion and virion entry.
Methods
Cells, virus and plasmids
All virus mutants derived from HSV-1 strain F (kindly provided by Y. Mettenleiter, Chicago, IL) were grown on African green monkey (Vero) cells in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal calf serum. For transient-complementation experiments, UL43 gene of HSV-1 F was cloned into pCDNA3 plasmid (Life Technologies-Invitrogen) and named pcDNA-HUL43.
Generation of recombinant viruses
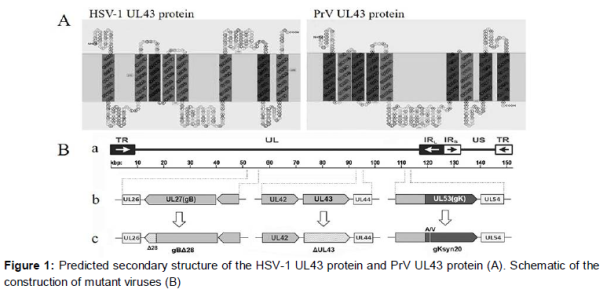
For generation of HSV-1 ΔUL43, the UL43 ORF was mutagenized with synthetic oligonucleotides HUL43-MUT for (5’-TCACGGCCAAGGCGCG GGcTGTGGGGCGTGACGGAACCAAGGATGACGACGATAAGTAGGG-3’) and HUL43-MUT rev (5’-TCCGTCACGCCCCACATAgCCCGCGCCTT GGCCGTGACAACCAATTAACCAATTCTGATTAG-3’), which mutates the initiation codon from ATG to CTG (shown in lowercase), and HSVΔUL43-seq-s (5’-CACAGAATTCGCAAGGC GTTGCTGTCG-3’) and HSVΔUL43-seq-as (5’-GCGGCTCGAGCGCTAAAAAGTTATTT-3’). Using these primers to amplify the AphA1 gene located on pEPkan-S, a PCR product was produced that contained the desired mutation and the kanamycin resistance gene. Mutagenesis was accomplished by using a markerless two-step Red recombination mutagenesis system [11] implemented on the HSV-1 (F) bacterial artificial chromosome (BAC) as described previously [12,13]. The desired HSV-1 ΔUL43 was isolated from non-fluorescent progeny virus plaques. The pHSV1-ΔUL43 was used as the backbone for construction of HSV1 gBΔ28/ΔUL43 and gKsyn20/ΔUL43 by introducing the designated mutations (B).
Preparation of antiserum
The HSV-1 L43 gene product is predicted to possess multiple membrane spanning domains (A). We ordered for recombinant membrane protein HSV-1 UL43 (Catalog: MBS1151851) which is His tagged from MyBioSource company. The 43-kDa His-UL43 recombinant protein was purified and used for immunization in a rabbit as described [14].
Virus purification
Vero cells were infected at 0.001 pfu per cell and incubated for 3 days. The culture medium was harvested and centrifuged at 2000 rpm (Beckman GH3.8) for 20 min to remove cell debris. Supernatant virions were sedimented by centrifugation for 1 h at 20,000 rpm (Beckman SW32 rotor) through a 40 % sucrose cushion in phosphate-buffered saline. The clear virus band was harvested and the virions were pelleted at 20,000 rpm for 2 h. The pellets were re-suspended in PBS.
Plaque morphology and one-step growth kinetics
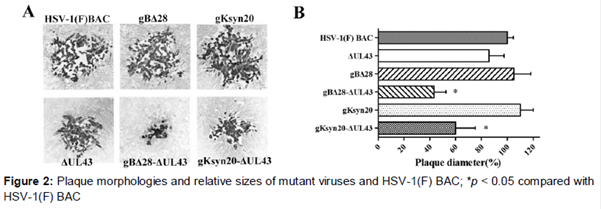
Cell monolayers were infected at 37 oC for 1 h and then overlaid with CMC (0.6 % carboxymethyl cellulose in DMEM + 2 % FCS; Sigma). After incubation at 37 oC for 48 h, cells were fixed with ice-cold methanol and plaques were visualized after immunohistochemical staining with polyclonal anti-HSV rabbit sera. At 24 h post transfection, cells were infected with mutant viruses lacking UL43 and incubated as described above. Average plaque sizes and standard deviations were determined and compared to the mean size of plaques induced by HSV-1 (F) BAC, which was set as 100 %.
To monitor one-step growth, Vero cells were infected with each virus at a multiplicity of infection (MOI) of 5 and incubated on ice for 1 h. Thereafter, the cells were incubated at 37 °C and virus was allowed to penetrate for 1 h at 37 °C. Any remaining extracellular virus was inactivated by low-pH treatment (pH 3.0), and incubation was continued at 37 °C. At 0, 4, 12, 24, 36, and 48 hpi, virus stocks were prepared and the virus progeny was titrated on Vero cells.
Quantification of cell-to-cell fusion
Subconfluent Vero cells in six-well plates were transfected with the plasmid containing T7 RNA polymerase gene (effector cells) or luciferase gene under the control of the T7 promoter (target cells) by using Lipofectamine 2000 (Invitrogen). As positive and negative controls, cells were transfected with both of the plasmids simultaneously and with pCAGGS empty vector plasmid, respectively. Twelve hours post transfection, effector and target cells were detached and seeded in a 24-well plate at a 1:1 ratio.
Twelve hours post seeding, the monolayers were infected with HSV-1 (F) BAC and mutant viruses at an MOI of 0.2. At 12 and 24 hpi, cells were washed with PBS and lysed with passive lysis buffer. Supernatants were collected and reacted with luciferase substrate (Promega, Madison, WI). The luminescence was measured with a TD-20/20 luminometer (Turner Designs).
SDS-PAGE and Western blot assay
Vero cells were infected with HSV-1 (F) BAC or mutant viruses at a MOI of 5. Cells were harvested at 24 hpi and lysed with NP-40 lysis buffer (Life Technologies-Novex). The collected samples were separated by SDS-PAGE (Tris-HEPES-SDS gradient 4 to 20 % gels) followed by electrophoretic transfer to nitrocellulose membrane and the presence of various proteins was determined using the primary antibodies described below. Goat anti-mouse secondary antibody conjugated with horseradish peroxidase (HRP) and enhanced chemiluminescence (ECL) substrate were used for detection purposes.
Primary antibodies used were: anti-HSV-1 gB, gD, gH, gL and ICP5 (VP5) monoclonal antibodies (MAbs) (Virusys), anti-HSV-1 gH MAb (Abcam), rabbit anti-HSV-1 UL43 polyclonal antibody (pAb), anti-tubulin alpha (AbD Serotec).
Analysis of membrane-associated proteins
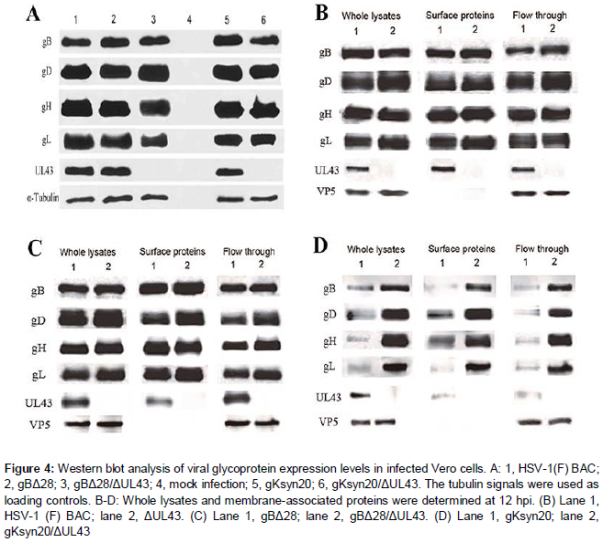
Biotinylation of cell surface proteins was used to identify membrane-associated proteins of infected Vero cells. Surface proteins of Vero cells infected with the indicated viruses were biotinylated at 12 hpi and isolated with a Pierce cell surface protein isolation kit (Thermo Scientific). The isolated proteins, whole lysates, and flow through were analyzed by Western blots with specific antibodies to gB, gD, gH, gL, UL43, and VP5.
Virus-penetration assay
Vero cell monolayers were infected at 4 oC for 1 h with approximately 250 pfu of HSV-1 (F) BAC and ΔUL43 mutant viruses. The medium was removed and cells were washed in ice-cold PBS and then incubated at 37 oC to allow virus penetration. Immediately thereafter (0 min) and at 30, 60, 120, and 180 min, unabsorbed virus was inactivated by acid wash. Samples without acid wash served as controls for input virus levels. The cells were overlaid with CMC and fixed with ice-cold methanol at 48 hpi. Plaque numbers were counted for each time point and to determine the entry kinetics, linear regression slopes from 0 to 120 min were calculated.
Statistical analysis
All data are expressed as arithmetic mean ± standard deviation (SD, n ≥ 3. Data were processed with GraphPad Prism 5.0, using one-way ANOVA, where appropriate. P < 0.05 was considered statistically significant.
Results
Recombinant HSV-1 viruses
To assess the effect of UL43 on virus-induced cell fusion, we constructed a set of recombinant viruses containing either the gBΔ28 or gKsyn20 mutation in the presence or absence of mutations lacking the UL43 gene by using the HSV-1 F genome cloned as a BAC. The set of mutant viruses that were constructed included the following: (i) gBΔ28, (ii) gBΔ28/ΔUL43, (iii) gKsyn20, and (iv) gKsyn20/ΔUL43 (B and 2).
Growth properties of HSV-1 UL43 mutants
Lack of UL43 expression resulted in significantly reduced levels of cell-to-cell fusion and decreased plaque sizes of syncytial mutant viruses (A). In contrast, gB- or gK-mediated syncytia formation and virus spread were restored to nearly HSV-1 (F) BAC levels (B). As expected, deletion of UL43 severely inhibited cell fusion induced by gBΔ28 or gKsyn20, exhibiting cell fusion levels lower than those produced by the HSV-1 (F) BAC (A, B). One-step growth studies revealed that in Vero cells all viruses replicated in similar manners, achieving viral titers approximately equal to those of the parental virus irrespective of the presence of the engineered mutations (C, D).
HSV-1 glycoproteins in infected cells
A lack of UL43 expression was confirmed via the inability of anti-UL43 antibody to detect the presence of UL43 protein in the mutant viruses (A). Overall, the expression levels of glycoproteins gB, gD, gH and gL were highly similar between the recombinant and HSV-1(F) BAC viruses, indicating that lack of UL43 did not affect the synthesis of viral glycoproteins.
Cell surface expression of viral glycoproteins
Deletion of UL43 did not affect overall expression levels of the viral glycoproteins gB, gD, gH and gL on HSV-1(F) BAC infected cell surfaces. Similar results were observed in the case of gBΔ28 virus (B-D).
HSV-1 UL43 is required for efficient virus entry
A significant delay in the rate of ΔUL43 virus entry was observed compared with the HSV-1(F) BAC (). These observations suggested that UL43 protein is required for wild-type like virion entry, and the reduced entry rate of virions lacking UL43 could at least partly account for the defects in cell-to-cell spread of this virus.
Discussion
HSV-1 enters cells predominantly via fusion of its viral envelope with cellular plasma membranes and can spread from infected to uninfected cells via virus-induced cell-to-cell fusion. Both membrane fusion phenomena are thought to be mediated by similar mechanisms involving glycoproteins gB, gD, gH, and gL, which form a functional complex that is required for both virus entry and virus-induced cell fusion. The purpose of this study was to investigate whether the UL43 protein is involved in these membrane fusion phenomena. In this report, these data demonstrate a functional role of UL43 protein in regulating the HSV-1 membrane fusion during virus-induced cell fusion and virion entry.
The UL43 protein is highly hydrophobic and has been predicted to span membranes at least eight times (A). Our detailed analysis of HSV-1 UL43 deletion mutants confirmed previous findings that the UL43 protein is not required for virus growth in cell culture [15]. The BMRF2 protein, which is the UL43 homologous protein in Epstein-Barr-Virus (EBV), is involved in attachment and entry into polarized epithelial tongue and nasopharyngeal cells by binding to cellular integrin [16]. By comparison, a lack of HSV-1 UL43 was reported to have little effect in a mouse infection model [15], arguing against an essential role of UL43 in virus infection in vivo. Thus, studies in a natural virus-host system using appropriate mutant viruses are needed to be performed to solve this problem.
Although deletion of UL43 did not abrogate HSV-1 replication, it significantly affected cell-to-cell spread and inhibited virus-induced cell fusion caused by a syncytial mutation in either gB or gK. The gBΔ28 mutation is the strongest gB syncytial mutation causing extensive cell fusion in tissue culture [17,18]. Unlike gB syncytial mutations, gKsyn20 mutations are known to cause extensive virus-induced cell fusion of all cell types, and gB syncytial mutations cause weaker fusion in a more limited repertoire of cells [19].
The experiments described herein have revealed that HSV-1 UL43 protein could be expressed on infected Vero cell surfaces and is required for virus-induced cell fusion. However, the PrV UL43 protein was suggested to inhibit membrane fusion in the transient co-expression system involving PrV envelope glycoproteins [10]. These findings seem to contradict results obtained herein with the recombinant viruses lacking UL43 gene. Interestingly, a similar result was observed for glycoprotein gM and gK, which inhibited cell fusion, caused by transient co-expression of gB, gD, gH, and gL [20-22] but were required for virus-induced cell fusion caused by syncytial mutations in gB [23]. These results indicate that UL43, as well as gM and gK, may regulate virus-induced cell fusion both positively and negatively depending on the presence of different sets of viral proteins.
Transient coexpression of gB, gD, gH, and gL is necessary and sufficient for the induction of cell-to-cell fusion [24]. In spite of these Davis-Poynter et al concluded that UL43 are dispensable for syncytium formation [23], the recombinant viruses lacking the UL43 gene were constructed in the presence of strong syncytial mutations in glycoprotein B (gB) or gK. Lack of UL43 did not appear to appreciably impact the overall expression level of these viral glycoproteins or their relative levels expressed on infected cell surfaces, suggesting that UL43 may indirectly or physically interact with one or more of the viral glycoproteins to regulate cell fusion. Furthermore, it is possible that the roles of UL43 protein, gM and gK are redundant and related since all of these proteins represent multiple membrane spanning proteins.
The effect of UL43 protein on the highly complicated virus-induced cell fusion machinery remains largely unexplored and will be the subject of future experiments. In this regard, these results support the hypothesis that fusion of the viral envelope with cell membranes is similar to virus-induced cell fusion. A number of viral proteins are involved in virus-induced cell fusion machinery.
Thus, it is conceivable that deletion of viral membrane proteins other than gB, gD, gH, and gL may affect viral entry, as has been shown here for UL43. Other membrane-associated proteins embedded in the viral envelope may exert site-specific regulation of membrane fusion phenomena mediated by fusogenic glycoproteins such as gB, one of which may be the optimal recognition of various receptors and entry into different types of cells.
Conclusion
Collectively, these results demonstrate an important role for HSV-1 UL43 protein in virus-induced cell fusion and efficient virus entry, although deletion of UL43 did not substantially affect virus replication in the cell culture. It is apparent that multiple functional interactions occur among viral glycoproteins and other envelope proteins that modulate membrane fusion phenomena in HSV-1 infections.
Declarations
Acknowledgement
References
Archives


News Updates