Supriyo Saha1 ![]() ,
Dilipkumar Pal2,
Sushil Kumar3
,
Dilipkumar Pal2,
Sushil Kumar3
For correspondence:- Supriyo Saha Email: supriyo9@gmail.com Tel:01356457512
Received: 17 July 2015 Accepted: 17 June 2016 Published: 31 July 2016
Citation: Saha S, Pal D, Kumar S. Design, synthesis and antiproliferative activity of hydroxyacetamide derivatives against HeLa cervical carcinoma cell and breast cancer cell line. Trop J Pharm Res 2016; 15(7):1401-1411 doi: 10.4314/tjpr.v15i7.8
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To design and develop a new series of histone deacetylase inhibitors (FP1 - FP12) and evaluate their inhibitory activity against hydroxyacetamide (HDAC) enzyme mixture-derived HeLa cervical carcinoma cell and MCF-7.
Methods: The designed molecules (FP1 - FP12) were docked using AUTODOCK 1.4.6. FP3 and FP8 showed higher interaction comparable to the prototypical HDACI. The designed series of 2-[[(3-Phenyl/substituted Phenyl-[4-{(4-(substituted phenyl)ethylidine-2-Phenyl-1,3-Imidazol-5-One}](-4H-1,2,4-triazol-5-yl)sulfanyl]-N-hydroxyacetamide derivatives (FP1-FP12) was synthesized by merging 2-[(4-amino-3-phenyl-4H- 1, 2, 4-triazol-5-yl) sulfanyl]-N-hydroxyacetamide and 2-{[4-amino-3-(2-hydroxyphenyl)-4H-1,2, 4-triazol-5-yl]sulfanyl}-N hydroxyacetamide derivatives with aromatic substituted oxazolone. The biological activity of the synthesized molecule (FP1-FP12) was evaluated against HDAC enzyme mixture-derived HeLa cervical carcinoma cell and breast cancer cell line (MCF-7).
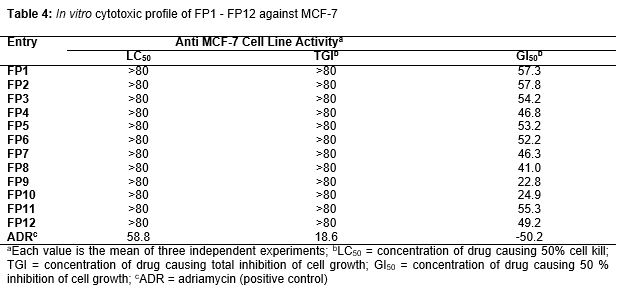
Results: HDAC inhibitory activity of FP10 showed higher IC50 (half-maximal concentration inhibitory activity) of 0.09 μM, whereas standard SAHA molecule showed IC50 of 0.057 μM. On the other hand, FP9 exhibited higher GI50 (50 % of maximal concentration that inhibited cell proliferation) of 22.8 μM against MCF-7 cell line, compared with the standard, adriamycin, with GI50 of (-) 50.2 μM.
Conclusion: Synthesis, spectral characterization, and evaluation of HDAC inhibition activity and in vitro anticancer evaluation of novel hydroxyacetamide derivatives against MCF-7 cell line have been achieved. The findings indicate the emergence of potentialanticancer compounds.
Introduction
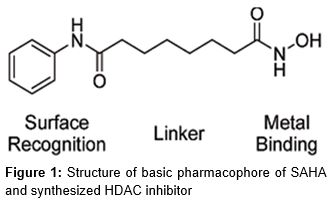
Histone deacetylase (HDAC) enzyme is one of the leading targets in the process of anticancer drug development. HDAC was divided into three distinct structural classes as class (I/II) zinc- dependent and class (111) NAD dependent [1]. These enzymes are a part of multiprotein complexes, catalyzing the removal of acetyl group from lysine residue on protein including histone. Suberoyl anilide hydroxamic acid was the primary important histone deacetylase inhibitor (HDACI) ().
Suberoyl anilide hydroxamic acid has three basic parts as surface recognition, linker and metal binding portions. HDAC inhibitors have shown to bind the active site and block the substrate access, causing a resultant accumulation of acetylated histones [2]. HDACIs inhibit tumor growth, cell differentiation and programmed cell death. HDACIs induce cancer cell cycle arrest, growth inhibition, differentiation, and programmed cell death [3]. HDACI induced cell cycle arrest and growth inhibition is usually correlated with transcriptional activation of p21WAF1/CIP1, p27KIP1, GADD45a [4] and inhibition of cyclin A, cyclin D and thymidylate synthetase [5]. The most important HDACI are the hydroxamic acids group followed by the benzamides, the cyclic tetra peptides, the carboxylic acids and the electrophilic ketones [6]. These observations prompted us to modify the linker portion to an imidazolo triazolated moiety and enacting the metal binding hydroxamic acid portion and molecular docking against 1T69, synthesize and check their in vitro HDAC inhibition, antiproliferative efficacy against MCF-7 cell line.
Methods
Molecular docking protocol and binding analysis
All computational studies were carried out using AUTODOCK 4.0.1. The geometry of HDAC-8 was extracted from the Brookhaven protein data bank (PDB entry code: 1T69) complexes with the irreversible inhibitor SAHA (Suberoyl Anilde Hydroxamic acid). All the residues within 20 Å core from SAHA were used to define the metal binding site. For the docking, a grid spacing of 0.375 Å and 126 × 90 × 90 number of points was used. The grid was centered on the mass center of the experimental bound SAHA coordinates. Autodock generated 10 possible binding conformations. A default protocol was applied, with an initial population of 150 randomly placed individuals, a maximum number of 2.5 x 105 energy evaluations, and a maximum number of 2.7 x 104 generations. A mutation rate of 0.02 and a crossover rate of 0.8 were used [7-10]. Cluster analysis of the docking results using root mean square deviation (RMSD) tolerance of 2 Ǻ.
Materials and equipment
Melting points were checked using open capillary method Veego Electronics Apparatus. The IR spectra for comparison of synthesized compounds were recorded on a Perkin Elmer (serial no: 78625) FTIR spectrophotometer. 1HNMR spectra were recorded on Bruker Avance DRX300 300MHz FTNMR spectrometer using DMSO-d6 as solvent. The chemical shifts were measured at δ units (reported as ppm) relative to TMS and signals are reported as s (singlet), d (doublet), t (triplet), q (quartet, m (multiple). Mass spectra were also recorded. All 1HNMR and Mass spectra was done by IICB, Kolkata and CDRI, Luck now. UV spectroscopy was done by the help of Shimadzu UV-1700 Spectroscopy. Elemental analysis was performed using a micro-analytical unit. All chemicals were procured from sigma Aldrich and Merck. All the reactions were routinely checked by precoated Merck thin layer chromatographic plate using toluene:methanol (9:1) as solvent system.
Chemistry
Synthesis of 2-chloro-N-hydroxyacetamide (III)
Methanol (12 mL) and 0.0336 M, 2.34 g of hydroxylamine hydrochloride were placed over a heated magnetic plate in a flask. The mixture was stirred for 5 min and added drop wise to a previously prepared methanol solution of 0.0501 M, 2.81 g potassium hydroxide. Then the resulting solution was cooled to room temperature and filtered. The filtrate was stored and used for the next step.
Then 25 ml of methanol was taken in a 250 ml beaker and stir over magnetic stirrer. Total synthetic setup was done under fuming cupboard. Pour 0.07 M, 5.6 ml of chloroacetyl chloride from dropping funnel onto the methanol and mixed it properly. Then hydroxylamine stock solution was added dropwise to the choloroacetyl chloride and stir for 2 h. The product was obtained by filtration. Filtrate was washed with diethyl ether and recrystallized from methanol.
Physical Nature: White Colored powder solid. Yield: 33.25 % M.p = 138 ˚C. Total synthetic procedure was report in Scheme 1.
General procedure for the preparation of 2-[(4-amino-3-phenyl/3-(2-hydroxy phenyl)-4H- 1, 2, 4-Triazol-5-yl) sulfanyl]-N-hydroxyaceta-mide (IX and XV)
4-amino-3-phenyl-4H-1, 2, 4-triazole-5-thiol (VIII) was synthesized from benzoic acid and 2-(4-amino-5-sulfanyl-4H-1, 2, 4-triazol-3-yl) phenol (XIV) from salicylic acid by adopting Reid Hindel Process. Both 4-amino-3-phenyl-4H-1, 2, 4-triazole-5-thiol (VIII) and 2-(4-amino-5-sulfanyl-4H-1, 2, 4-triazol-3-yl) phenol (XIV) were stirred with 2-chloro-N-hydroxyacetamide (III) in dimethyl formamide (DMF) solution for 1h respectively to achieve compound IX and XV. The general procedure for the synthesis of IX and XV was reported in Scheme 2 and 3.
General procedure for the preparation of (4E)-4-[(2-) methylidene]-2-phenyl-1,3-oxazol-5(4H)-one (XVIIIa-f)
An amount of 0.022 M substituted aromatic aldehyde (benzaldehyde, salicylaldehyde, 4-methoxy benzaldehyde, anisaldehyde, furfuraldehyde, cinnamaldehyde), 1.8 g of 0.022 M sodium acetate and 4.0 g of 0.022 M hippuric acid were suspended in 0.066 M glacial acetic acid solution. The mixture was refluxed for 2 h under water bath.
After cooling the mixture, add 10 mL of ethanol into the solution and kept for overnight at below 5 oC. The obtained precipitation was filtered and washed the filter cake with ethanol followed by drying under vacuum. Synthesis was performed as per Scheme 4 and result reported in .
General procedure for the preparation of 2-[[(3- substituted phenyl-[4-{(4-(substituted phenyl) ethylidine-2-Phenyl-1,3-Imidazol-5-One}](-4H-1,2,4-triazol-5-yl)sulfanyl]-N-hydroxyacetamide (FP1 - 12)
Equimolar concentration (0.01 M) of (4E)-4-[(substituted phenyl/methyl) methylidene]-2-phenyl-1,3-oxazol-5(4H)-one and 0.01M of 2-[(4-amino-3-phenyl-4H- 1, 2, 4-triazol-5-yl) sulfanyl]-N-hydroxyacetamide/2-{[4-amino-3-(2-hydroxy phenyl)-4H-1, 2, 4-triazol-5-yl]sulfanyl}-N-hydroxyacetamide was added to a 250 mL RBF and refluxed for 5 h at 150 oC under oil bath using 0.01 M pyridine and 0.01 M Zeolite (Y-H) as catalyst. After completion of the reaction, the excess of pyridine was distilled off. The resultant solution was cooled and poured into crushed ice and hydrochloric acid mixture. The obtained product was filtered and recrystallized from ethanol. Total synthetic procedure from FP1-12 was reported in Schemes 5 and 6.
In vitro HDAC inhibition assay
In vitro fluorescent histone deacetylase assay: HDAC inhibition assays were performed using the HDAC fluorescent activity assay kit. HeLa cell nuclear extract which contains a number of HDAC isozymes and other nuclear factors, was used as the source of HDAC activity. The final substrate concentration in the assay mixture was 50 µM. The reaction was allowed to proceed for 10 min at room temperature before a stop solution was added. Test compounds were prepared as 20 mM stock solutions in DMSO and stored at -70 oC. DMSO had no significant effect on the activity of this assay at concentrations up to 5 % with the final DMSO concentration in the assays of not more than 2 %. Assays were performed in white polystyrene 96-well half-area assay plates and measured on a Wallac 1420 fluorescent plate reader with an excitation wavelength of 355 nm, an emission wavelength of 460 nm, and a one sec signal averaging time [13].
Evaluation of antiproliferative activity on human breast cancer cell line (MCF-7)
Antiproliferative activity of FP1-FP12 was evaluated using Sulphorodamine B (SRB) assay method on MCF-7 cell line [14]. Cultures fixed with trichloroacetic acid were stained for 30 min with 0.4 % (w/v) sulforhodamine B (SRB) dissolved in 1 % acetic acid. Unbound dye was removed by four washes with 1 % acetic acid, and protein-bound dye was extracted with 10 mM unbuffered Tris base [tris hydroxymethyl) aminomethane] for determination of optical density in a computer-interfaced, 96-well micro titer plate reader. SRB assay results were linear with the number of cells and with values for cellular protein measured by both Lowry and Bradford assays at densities ranging from sparse sub-confluence to multilayered supraconfluence. The signal-to-noise ratio at 564 nm was approximately 1.5 with 1,000 cells per well.
The sensitivity of SRB assay compared favorably with sensitivities of several fluorescence assays and was superior to those of both Lowry and Bradford assays and to those of 20 other visible dyes. SRB assay provides a colorimetric end point that is nondestructive, indefinitely stable, and visible to the naked eye. It provides a sensitive measure of drug-induced cytotoxicity, is useful in quantitating clonogenicity, and is well suited to high-volume, automated drug screening. SRB fluoresces strongly with laser excitation at 488 nm and can be measured quantitatively at the single-cell level by static fluorescence cytometry.
Statistical analysis
All the data were subjected to one-way ANOVA followed by Dunnett’s test. Statistical analysis was carried out using GraphPad Prism software 5.0. Statistically significant difference was set at p < 0.05.
Results
Molecular docking
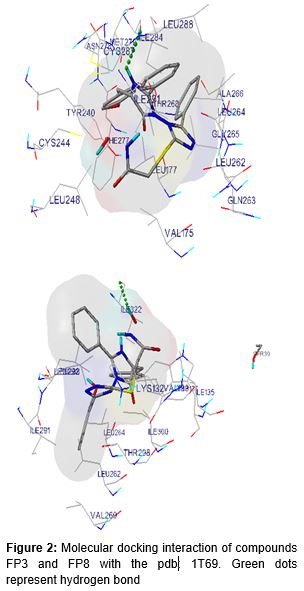
Energy minimized conformer with best Dock scores was considered for the identification of interacting amino acid residues with ligands. All the binding interactions were tabulated and diagrammatized in the ().
Synthesized compounds
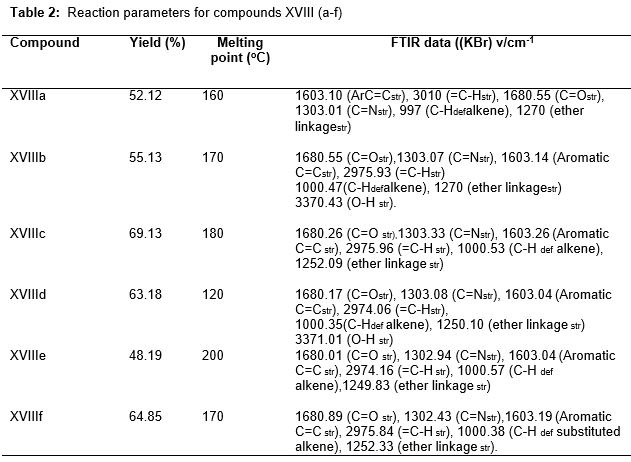
Twelve new hydroxyacetamide derivatives (FP1 -FP12) were prepared by the condensation of different oxazolone (XVIIIa-f) and appropriately substituted triazolated hydroxyacetamide (IX and XV) using reported method as per Schemes 5 and 6 [11,12]. The compounds were re-crystallized using ethanol. All the compounds were characterized by detailed spectroscopic (IR, 1H NMR, Mass) analyses.
FP1: Yield= 30.42%, M. p= 200˚C. FT-IR (KBr) v/ cm-1 769.60 (mono substituted aromatic ring def), 1610.67 (C=O str), 1351.88 (–OH def), 1501.17 (–NH def), 1558.15 (C=N str), 2951.81(C=S str), 1306.38 (N-N=C str) 1573.58 (nitrogen containing heterocyclic ring in combination of C=C). 1H NMR DMSO (300 MHz) δ ppm: 7.31-8.16 (m, 15H, Ar-H), 7.86 (s, 1H, NH), 3.33 (1H aliphatic CH2), δ 2.50 (1Hwas observed for hydroxamic OH proton). C26H20N6O3S: calculated C: 62.90%, H: 4.03%, N: 16.9%. C26H20N6O3S: found: C: 61.30%, H: 4.02%, N: 16.5%. (M+1) 496.12 (EIMS).
FP2: Yield= 28.55%, M. p= 210˚C. FT-IR (KBr) v/ cm-1 769.70 (mono substituted aromatic ring def), 1610.51 (C=O str), 1351.64 C=N str as well as –OH def), 1500.50 (–NH def), 2952.52 (C=S str), 1306.06 (N-N=C str) 1573.21(nitrogen containing heterocyclic ring in combination of C=Cstr), 1558.03 (C=N str). 1H NMR DMSO (300 MHz) δ ppm: δ 7.87 (s, 1H, N-H), δ 7.61 (m, Ar-H), δ 3.32 (1 H aliphatic CH2), δ 2.50 (s, 1H, OH). C26H20N6O4S: calculated C: 60.90%, H: 3.92%, N: 16.47%. C26H20N6O4S: found: C: 60.14%, H: 3.89%, N: 16.4%. (M+1) 510.32 (EIMS).
FP3: Yield= 43.84%, M. p= 158˚C. FT-IR (KBr) v/ cm-1 843.26 (para substituted aromatic ring def), 1610.42 (C=O str), 1351.56 (C=N str), 1501.43 (–NH def), 2928.51 (C=S str), 1295.23 (N-N=C str), 1558.03 (C=N str), 1265.28 (Ar-OCH3), 3102 (NH str), 3280.51 (OHstr). 1H NMR DMSO (300 MHz) δ ppm: δ 7.87 (s, 1H, N-H), δ 7.61 (m, 14H, Ar-H), δ 3.38 (d, 2H aliphatic CH2), δ 2.50 (s, 1H, OH), 3.93 (t, 3H, OCH3 at 4th position of aromatic ring). C27H22N6O4S: calculated C: 61.53%, H: 4.18%, N: 15.96%. C27H22N6O4S: found C: 60.50%, H: 4.10%, N: 15.50%. (M+1) 526.32 (EIMS) and 179.0266 the main fragmented portion.
FP4: Yield= 35.21%, M. p=155˚C. FT-IR (KBr) v/ cm-1 802.00 (para substituted aromatic ring def), 1609.38 (C=O str), 1351.56 (C=N str), 1501.43 (–NH def), 2927.45(C=S str), 1308.14 (N-N=C str), 1573.91(nitrogen containing heterocyclic ring in combination of C=C), 1558.03 (C=N str), 1266 (Ar-OCH3). 1H NMR DMSO (300 MHz) δ ppm: δ 7.86 (s, 1H, N-H), δ 7.45 (m, 14H, Ar-H), δ 3.38 (d, 2H aliphatic CH2), δ 2.50 (s, 1H, OH), 3.91 (t, 3H, OCH3 at the C-3 position of aromatic ring), 8.82 (s, 1H, Phenolic OH). C27H22N6O5S: calculated C: 59.71%, H: 3.93%, N: 15.02%. C27H22N6O5S: found C: 59.574%, H: 3.85%, N: 14.88%. (M+1) 559.0299 (EIMS) and the main fragmented portion is 179.0266.
FP5: Yield= 23.21%, M. p=205˚C. FT-IR (KBr) v/ cm-1 790.37 (mono substituted aromatic ring def), 1601.61 (C=O str), 1340.57 (C=N str), 1308.12 (N-N=C str), 1266.29 (etheric linkage). 1H NMR DMSO (300 MHz) δ ppm: δ 7.85 (s, 1H, N-H), δ 7.57 (m, 10H, Ar-H), δ 3.35 (d, 2H aliphatic CH2), δ 2.50 (s, 1H, OH), 7.64 (s, 1H, furan H). C24H18N6O4S: calculated C: 59.19%, H: 3.70%, N: 17.28%. C24H18N6O4S: found C: 58.35%, H: 3.63%, N: 16.96%. (M+1) 486.02 (EIMS) and 179.0268.
FP6: Yield= 25.96%, M.p=188˚C. FT-IR (KBr) v/ cm-1 790.374 (mono substituted aromatic ring def), 1609.98 (C=O str), 1351.19 (C=N str), 1307.49 (N-N=C str), 1266.29 (etheric linkage). 1H NMR DMSO (300 MHz) δ ppm: δ 7.86 (s, 1H, N-H), δ 7.57 (m, 10H, Ar-H), δ 3.32 (d, 2H aliphatic CH2), δ 2.50 (s, 1H, OH), 3.44 (s, 1H, CH3). C21H18N6O3S: calculated C: 58.00%, H: 4.15%, N: 19.39%. C21H18N6O3S: found C: 57.613%, H: 4.02%, N: 18.95%. (M+1) 433.2019 (EIMS).
FP7: Yield= 33.12%, M.p= 190˚C. FT-IR (KBr) v/ cm-1 749.59 (mono substituted aromatic ring def), 1592.68 (C=O str), 1384.68 C=N str as well as –OH def), 1487.21 (–NH def), 2932.62 (C=S str), 1291.72 (N-N=C str). 1H NMR DMSO (300 MHz) δ ppm: δ 7.84 (s, 1H, N-H), δ 7.23 (m, Ar-H), δ 3.91 (1 H aliphatic CH2), δ 2.72 (d, 1H, Ar-OH) δ 2.50 (s, 1H, OH), C26H20N6O4S: calculated C: 60.94%, H: 3.91%, N: 16.40%. C26H20N6O4S: found C: 60.13%, H: 3.85%, N: 15.85%. (M+1) 512.58 (EIMS).
FP8: Yield= 27.65%, M. p= 205˚C. FT-IR (KBr) v/ cm-1 750.87 (mono substituted aromatic ring def), 1592.37 (C=O str), 1384.68 (C=N str as well as –OH def), 1487.40 (–NH def), 2932.21 (C=S str), 1291.85 (N-N=C str). 1H NMR DMSO (300 MHz) δ ppm: δ 7.81 (s, 1H, N-H), δ 7.29 (m, Ar-H), δ 3.91 (1 H aliphatic CH2), δ 2.50 (d, 1H, Ar-OH) δ 2.50 (s, 1H, OH). C26H20N6O5S: calculated C: 59.21%, H: 3.78%, N: 15.90%. C26H20N6O5S: found C: 60.143%, H: 3.65%, N: 15.58%. (M+1) 528.52 (EIMS).
FP9: Yield= 33.64%, M. p= 185˚C. FT-IR (KBr) v/ cm-1 763.44 (mono substituted aromatic ring def), 1593.84 (C=O str), 1384.65 (C=N str as well as –OH def), 1512.01 (–NH def), 2933.89 (C=S str), 1321.24 (N-N=C str). 1H NMR DMSO (300 MHz) δ ppm: δ 7.79 (s, 1H, N-H), δ 7.39 (m, Ar-H), δ 3.81 (1 H aliphatic CH2), δ 2.66 (d, 1H, Ar-OH) δ 2.50 (s, 1H, OH). C27H20N6O5S: calculated C: 59.77%, H: 3.69%, N: 15.49%. C27H20N6O5S: found C: 59.90%, H: 3.52%, N: 15.04%. (M+1) 542.54 (EIMS).
FP10: Yield= 31.51%, M. p= 192˚C. FT-IR (KBr) v/ cm-1 807.57 (para substituted aromatic ring def), 1644.98 (C=O str), 1384.71 (C=N str), 1501.04 (–NH def), 2932.40 (C=S str), 1332.97 (N-N=C str), 1594.13 (nitrogen containing heterocyclic ring in combination of C=C), 1558.03 (C=N str), 1157.10 (Ar-OCH3). 1H NMR DMSO (300 MHz) δ ppm: δ 7.86 (s, 1H, N-H), δ 7.14 (m, 14H, Ar-H), δ 3.31 (d, 2H aliphatic CH2), δ 2.50 (s, 1H, OH), 3.31 (t, 3H, OCH3 at 3rd position of aromatic ring), δ 2.50 (s, 1H, OH). C27H22N6O6S: calculated C: 57.96%, H: 3.94% N: 15.05%. C27H22N6O6S: found C: 58.13%, H: 4.02%, N: 14.85%. (M+1) 558.84 (EIMS).
FP11: Yield= 28.18%, M. p=212˚C. FT-IR (KBr) v/ cm-1 1807.41 (mono substituted aromatic ring def), 1644.54 (C=O str), 1329.34 (C=N str), 1308.12 (N-N=C str), 1266.29 (etheric linkage). 1H NMR DMSO (300 MHz) δ ppm: δ 7.58 (s, 1H, N-H), δ 7.12 (m, 10H, Ar-H), δ 3.31 (d, 2H aliphatic CH2), δ 2.50 (s, 1H, OH), 8.11 (s, 1H, furan H). C24H18N6O5S: calculated C: 57.31%, H: 3.58%, N: 16.73%. C24H18N6O5S: found C: 57.30%, H: 3.45%, N: 15.98%. (M+1) 502.90 (EIMS).
FP12: Yield= 45.23%, M. p=188˚C. FT-IR (KBr) v/ cm-1 807.48 (mono substituted aromatic ring def), 1644.98 (C=O str), 1331.87 (C=N str), 1308.76 (N-N=C str), 1266.65 (etheric linkage). 1H NMR DMSO (300 MHz) δ ppm: δ 7.859 (s, 1H, N-H), δ 7.138 (m, 15H, Ar-H), δ 3.311 (d, 2H aliphatic CH2), δ 2.500 (s, 1H, OH), δ 7.480, 5.265 (d, 1H, ethylene group of cinnamaldehyde). C28H22N6O4S: calculated C: 62.39%, H: 4.08%, N: 15.61%, C28H22N6O4S: found C: 62.85%, H: 3.92%, N: 14.85%. (M+1) 538.65 (EIMS).
In vitro HDAC inhibition
The in vitro HDAC inhibition activities of compounds FP1 - 12 are reported in terms of % inhibitory concentration (IC50). All the results were presented in and .
Antiproliferative activity against MCF-7 cell line
The in vitro cytotoxic activity of compounds FP1 - 12 are reported in terms of % GI50 and the data are shown in .
Discussion
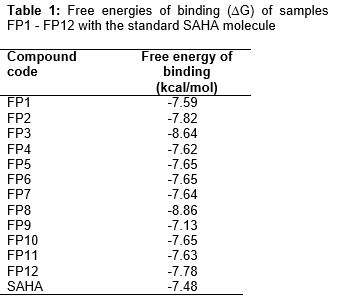
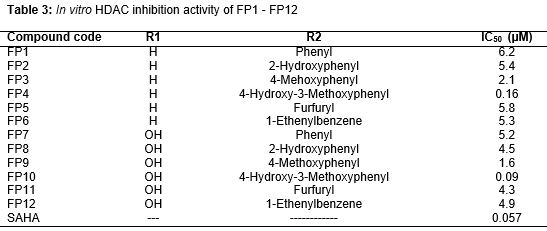
Docking result suggested that compound FP3, FP8 have shown a significant favorable free energy of binding (-) 8.64 kcal/mol and (-) 8.86 Kcal/mol which seems to be much close to that of the reference standard SAHA with -7.48 kcal/mol. In the showed that the surrounding residues of FP3 the surrounded residues were VAL 175, LEU 262, GLY 265, LEU 264, TYR 240, ILE 284, THR 268 and in the case of FP8 the surrounding residues were ALA 38, LEU 31, TRP 14, PHE 152, LYS 33, SER 39, ARG 37 which was subsequently present in the receptor voxel. HDAC8 receptor is a metal activated enzyme which consists of Zn 378 residue in its active site and in all the cases the docked molecules are present within a suitable range. All the synthesized molecules are characterized by FTIR, 1H NMR, elemental Analysis, and mass Spectrophotometer. As per the spectral data of compound FP1-FP6, FTIR and 1H NMR showed presence of characteristic peak around 1600 cm-1, 3100 cm-1, 3300 cm-1 due to C=O, -OH, -NH stretching, around 3000 cm-1 for phenyl group, 1380 cm-1 for the etheric linkage and δ 2.500 ppm, around 7.200 ppm, around 7.800 ppm, around 3.38 ppm due to hydroxamic OH, Aromatic –H, -NH, -CH2 group respectively but the presence of para amino group of the triazole moiety δ 5.793 ppm is missing the final set of compound. In the case of spectral data of compound FP7-FP12, as per the 1H NMR absences of para amino group of the triazole moiety δ 5.622 ppm. Further, the structures were established by mass spectra data accordance to their molecular formula. The activity of new compounds FP1 - FP12 against HDAC enzyme mixture derived HeLa cervical carcinoma cells reported in suggest that compounds (FP10, FP4, FP9) were showed apparently higher IC50 value as 0.09 μM, 0.16 μM, 1.6 μM, 22.8 μM and 24.9 μM respectively as comparison to the standard SAHA molecule with 0.057 μM, among the result only FP10, FP9, FP4 were non-significantly varied from standard molecule data. However in the case of activity of new compounds FP1 - FP12 against MCF-7 cell line reported in only FP9 showed relative GI50 Value with 22.8 μM as comparison to control Adriamycin (ADR) with GI50 value - 50.2 μM, among the result only FP9 was non-significantly varied from standard molecule data.
Conclusion
Hydroxyacetamide derivatives have been successfully synthesized and characterized by FTIR, NMR, elemental analysis and mass spectrometry. They exhibited inhibitory activity against HDAC enzyme mixture-derived HeLa cervical carcinoma cells and MCF-7 cell line. Further investigations are required the full activity profile and toxicological properties of the derivatives.
Declarations
Acknowledgement
References
Archives


News Updates