Abdallah Ahmed Elbakkoush1,
Suleman Atique1,3,
Anas Khaleel2,
Chien-Tsai Liu1 ![]()
For correspondence:- Chien-Tsai Liu Email: ctliu@tmu.edu.tw
Received: 19 April 2016 Accepted: 15 August 2016 Published: 30 September 2016
Citation: Elbakkoush AA, Atique S, Khaleel A, Liu C. Pathway analysis for identification of potential biomarkers in severe cutaneous drug hypersensitivity reactions. Trop J Pharm Res 2016; 15(9):1839-1845 doi: 10.4314/tjpr.v15i9.4
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To construct a cluster model or a gene signature for Stevens-Johnson syndrome (SJS) using pathways analysis in order to identify some potential biomarkers that may be used for early detection of SJS and epidermal necrolysis (TEN) manifestations.
Methods: Gene ex
Results: Out of 193 genes, only 91 were used (after removing the ambiguous and duplicated genes) for topological analysis. It was found by geneMANIA database search that majority of these genes were co-expressed yielding 84.63 % co-ex
Conclusion: Analysis of differential gene ex
Introduction
Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are severe and rare cutaneous adverse reactions (SCAR) manifestations. It is believed that they are caused by certain drugs which may consequently lead to significant morbidity and mortality worldwide [1]. Although, effects of such manifestations are not localized, yet skin is the vulnerable tissue for such reactions. SJS and TEN are considered the most serious and fatal skin diseases among cutaneous drug-induced hypersensitivity adverse reactions [2]. SJS and TEN are immune mediated hypersensitivity reactions that involve the skin and mucous membranes. They are distinguished by bullous formation followed by epidermal detachment that eventually affects mucous membranes [3]. Currently, more than 5 % of the patients with SJS and 30 % of the patients with TEN die [4]. However, the underlying mechanism leading to such death have not been discovered yet.
T-cell pathways involvement was reported to be associated with TEN reaction over a decade ago, in addition to the recently reported Granulysin and natural killer cells inhibitory receptors [5-7]. Although these mechanisms were useful to decipher some of the largely unknown mechanisms underlying such a complex disease, yet there must be further predictors and biomarkers on the genetic level. Moreover, various studies have shown that several cytokines might play role in the apoptosis or the cytokine inflammations in SJS and TEN. Among these recently discovered cytokines are the interleukin (IL) IL-6, IL-8 and monocyte chemo-attractant protein-1 (MCP-1). All of these were upregulated in such manifestations [8].These cytokines and others are important cellular regulators and mobilizers of cells engaged in innate as well as adaptive inflammatory immunity which has interlocking factors to both SJS and TEN [9,10]. Cytokines are released by various cells in the body, usually in response to an activating stimulus, and they induce responses by binding to specific receptors on the surface of target cells [11]. Levels of pro-inflammatory cytokines were significantly higher in SJS and TEN patient samples than the healthy controls [12].
Main objective of the study is to establish relevant biomarkers for SJS and TEN early detection using the genetic mapping of DEG SJS and TEN patients’ samples. Protein-protein interaction network and pathways analysis tools along with other online databases and software not only provide information about these specific DEG proteins in severe cutaneous adverse reaction, but also pave the path towards determination of potential early detection biomarkers.
Methods
Gene set array
Gene expression profiles of GSE12829 were downloaded from Gene Expression Omnibus (GEO) database that has been investigated previously by Bellon et al [13]. The samples of twenty three patients suffering from cutaneous drug-induced hypersensitivity reactions were matched to eight healthy controls who were using the same medications. A total of 193 genes were found with distinct expressions in comparison between healthy controls and patients samples. GEO2R technique [14], (which is already available on Gene Expression Omnibus) was used to acquire the relevant gene list. Thereafter, these genes were loaded to geneMANIA database [15] to remove ambiguous and duplicate genes.
Database search
Gene expression profiles were characterized using DAVID (Database for Annotation, Visualization and Integrated Discovery). DAVID web tool provides a comprehensive set of functional annotation tools for researchers to understand biological meaning behind large list of genes [16]. In this study, the DAVID tool was used to annotate the function of our genes list. It lead to the selection of the Gene Ontology (GO) terms with adjusted p-value less than 0.05 and count larger than 5.
The STRING (Search Tool for the Retrieval of Interacting Genes) version 9.1 [17], database provides both experimental and predicted interaction information. Version 9.1 of STRING covers more than 1100 completely sequenced organisms. To identify the interactive relationships among target genes or other genes, genes list was mapped to STRING. GeneMANIA was again used to figure out genes that have protein and genetic interactions, pathways, co-expression, or co-localization [15]. It was also utilized to generate the relevant network with different scores.
The genes were then analyzed and characterized in KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway categories [18] by the GENECODIS software [19]. REACTOME is a bioinformatics tool for the visualization, interpretation and analysis of pathway knowledge to support basic research, genome analysis, modeling, systems biology and education [20].
Results
GeneMANIA analysis yielded 9-core nodes genes network
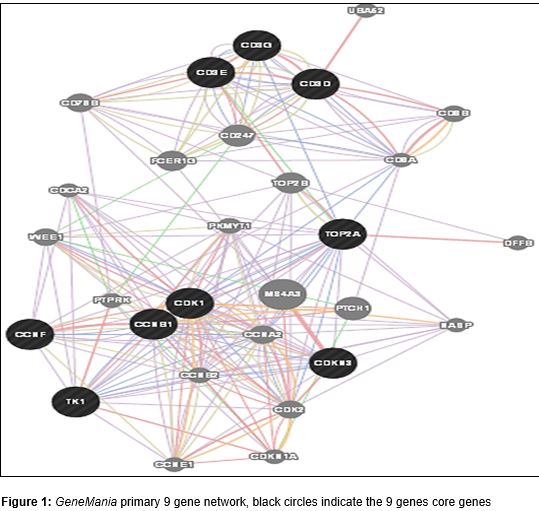
In this study, multiple databases approaches has been used to identify the pathways and network trends for the produced gene list. Primarily, majority of these genes were co-expressed together 84.63 % co-expression, and 14.33 % were in physical interaction. GeneMANIA database [15] search yielded only < 1% for the pathway and genetic interactions with values of 0.97 % and 0.06 %, respectively. The 9 nodes gene network was generated by GeneMania tool [15], as shown in .
Main protein-protein interaction networks generated by string (search tool for retrieval of interacting genes) version 9.1 [17]
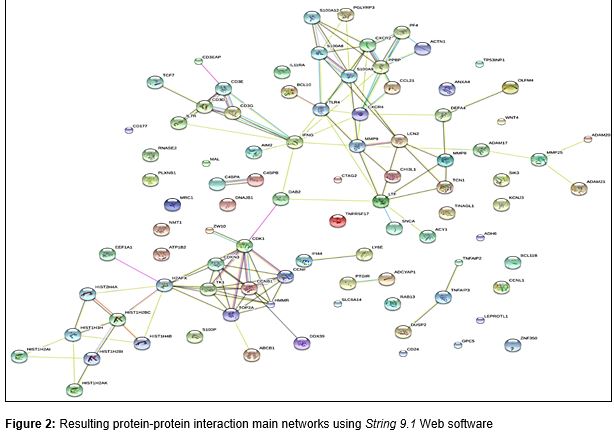
The protein-protein interaction network for our gene list, was identified using STRING (Search Tool for Retrieval of Interacting Genes) version 9.1 [17]. The result was a generic multiple interacting proteins network, as shown in .
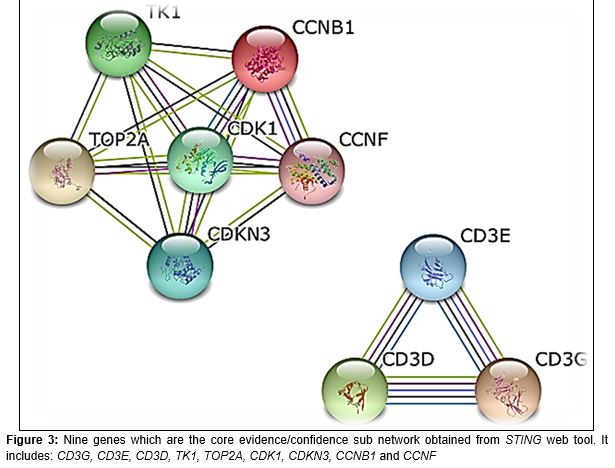
After further analysis nine genes with two cluster networks (where most of interacting protein in SJS and TEN present) were focused, and those interacting proteins sub-network (clusters) are shown in .
REACTOME pathways analysis results showed 13 gene clusters mainly in immune and signaling pathways
A set of 92 gene was applied to REACTOME pathways database [20]. The results reveal a 13-gene cluster that showed that the major pathways were related to immune system 54 (4 %), signal transduction 76 (4 %) and disease pathways 52 (5%).
DAVID; database for annotation, visualization and integrated discovery result is agreeing with REACTOME database
DAVID tool [16] results were also in accordance with the previous REACTOME output. Previous reports have shown T-cell mediated biological processes. DAVID results for the 13 gene were all targeting T-cell namely alpha-beta T-cell receptor complex, T-cell receptor complex, T-cell activation and phosphorylated immune signaling motifs (ITAMS).
Protein-protein interaction mainly participated in the biological processes which are related with immune system
GENECODIS software yielded condensed ten signaling pathways mainly in immune system
The genes were then analyzed and characterized by the GENECODIS program [19]. Ten significantly enriched signaling pathways are shown in descending order of the number of genes contained in each pathway, as explained in .
Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis gave two main modules consisting of cytokine-cytokine receptor and T-cell receptor pathways
The results of KEGG pathway enrichment analysis revealed that most of the genes in 2 modules exist in cytokine-cytokine receptor interaction and T cell receptor signaling pathway. Please refer to supplementary data S5 for further clarification.
Most of these DEG genes were found to be differentially expressed during the early phase of cutaneous drug-induced delayed hypersensitivity reactions. Furthermore, 9 genes were identified with distinct cluster expression and interaction patterns during the acute phase.
Discussion
This study have shown that specific gene cluster can be used not only for detection of early incidence of SJS and TEN but also to predict clinical prognosis in the same disease setting. Genes and pathways that were associated with incidence of SJS and TEN have been identified via this research. Moreover, it has also been identified that 9 genes signature in SJS and TEN which include: CD3G, CD3E, CD3D, TK1, TOP2A, CDK1, CDKN3, CCNB1, and CCNF. The full name of gene as follows: CD3G, T-cell surface glycoprotein CD3 gamma chain. CD3E, CD3e molecule, epsilon. CD3D, T-cell surface glycoprotein CD3 delta chain. TK1, Thymidine kinase 1. TOP2A, DNA topoisomerase 2-alpha. CDK1, Cyclin-dependent kinase 1. CDKN3, Cyclin-dependent kinase inhibitor 3. CCNB1, G2/mitotic-specific cyclin-B1. CCNF, G2/mitotic-specific cyclin-F.
Up till now, the pathogenesis of SJS/TEN is yet to be fully elucidated. STS and TEN are complex disease forms and could cause certain fatalities. The current study has deployed many online research engines and databases to analyze the differentially expressed genes in these clinical manifestations. geneMANIA results show that many of the inquired genes were ambiguous. However, the majority of these genes have evident role in immune and hypersensitivity reaction mechanisms [21]. Genes pathways in cytokine-cytokine receptor interaction and T-cell signaling shown by KEGG pathway analysis proved that these clusters are directly linked to SJS and TEN disease[18], and then the same has been confirmed via GENECODIS web tool as well [19]. Ten genes showing significantly enriched signaling pathways are shown in descending order of the number of genes contained in each pathway.
The first candidate pathway was cytokine-cytokine receptor interaction which has been validated in many reports to be associated with SJS and TEN manifestations. Previously published studies have shown that several cytokines might play an important role in the apoptosis or cytokine flooded inflammation [8,22,23]. Among these recently discovered cytokines are the interleukin (IL), IL-6, IL-8 and monocyte chemo-attractant protein-1 (MCP-1) and all were reported to be unregulated in SJS and TEN [8].
Cytokine-cytokine receptor interaction is the most significant pathway in KEGG and GENCODIS analysis. It is not unusual to find cytokines in SJS and TEN as they are crucial cellular regulators and mobilizers of cells engaged in innate as well as adaptive inflammatory host defense, cell growth, and differentiation aimed at the restoration of homeostasis. Cytokines are released by various cells in the body in response to an activating stimulus, and they induce response by binding to specific receptors on the surface of target cells in case of immunological response as well as in SJS and TEN disease pathophysiology [24].
Previous report by Miyagawa et al [25] state that expression pattern with lower expression of genes coding T cell-specific proteins and high expression of cell cycle-related genes and genes encoding for inflammatory related mediators have been shown [13].
This study extracted the gene set from Bellon et al which has been previously reported as well [13]. The drugs that cause the SJS were Clindamycin®, Spiramycin®, Statin, Phenytoin® and Allopurinol® [13]. These were the trigger of SJS manifestation and from where this gene expressions data come from. Meanwhile, Carbamazepine, furosemide, Erythromycin and paracetamol were more associated with TEN patients in the same cohort [13]. However, there are many other drugs that have shown substantial effects towards provoking SJS and TEN clinical reactions. For instance, in a study conducted by Barvaliya et al [26] it was found that antimicrobials were 50 % associated with these kind of reactions. NSAID’s and anti-seizure drugs were also associated and caused 22.41 % and 18.96 % of SJS and TEN cases respectively. According to the mentioned study Nevirapine was the most common drug in this regards which had an association of 28.12 %. They also figured out that anti-seizure drugs are mostly associated with such kind of adverse reactions.
Earlier data reports confirm that Han-Chinese subjects who have the HLA-B*1502 allele are highly prone to high risk of SJS and TEN after taking Carbamazepine medication. This allele however has higher prevalence in Asian and especially East Asian populations, including Han Chinese, Filipino, Malay, Indian, and Thai [27]. This proposes importance of gene loci in the investigation of SJS and TEN causal and association studies.
Conclusion
This research highlights 9 genes associated with the early incidence of SJS and TEN. In this study, pathways related gene cluster has also been identified and a model to predict SJS and TEN early incidences has been developed. These findings can be used in assisting and supporting clinical prognosis and diagnostic biomarkers for further investigation and future therapeutic discoveries. It can be suggested that SJS and TEN have certain potential link to genetic grounds which might be helpful for earlier diagnosis of such reactions to avoid more complications leading towards better clinical outcomes.
References
Archives


News Updates