Weihua Zheng,
Ying Wu ![]() ,
Wenyan Huang
,
Wenyan Huang
For correspondence:- Ying Wu Email: childhospsh@hotmail.com Tel:+2162474880
Received: 6 July 2016 Accepted: 24 October 2016 Published: 29 November 2016
Citation: Zheng W, Wu Y, Huang W. A case report on ATP6V0A4 gene mutation: Forecast of familial deafness by genetic investigation in a patient with autosomal recessive distal renal tubular acidosis. Trop J Pharm Res 2016; 15(11):2527-2530 doi: 10.4314/tjpr.v15i11.30
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
The autosomal recessive form of distal renal tubular acidosis (dRTA), a condition associated with the systemic accumulation of acid owing to its reduced elimination through kidneys, is caused by ATP6V0A4 mutation, which is typically related to either late-onset sensorineural hearing loss (SNHL) or normal hearing. This article reports dRTA in seven year old boy, born to a Chinese couple who have family history of deafness. The patient does not have hearing impairment. ATP6V0A4 gene sequencing demonstrated that there were 2 heterozygous mutations, c.1376C>T and c.1029+5G>A, in gene ATP6V0A4. c.1376C > T (p.Ser459Phe) is a kind of missense mutation in gene ATP6V0A4. c.1029+5G>A is a kind of intragenic mutation near the cutting area of gene ATP6V0A4. ATP6V0A4 gene mutation study substantiated the autosomal recessive dRTA without hearing impairment in the patient. This case report emphasizes the significance of early diagnosis and genetic screening of recessive forms of dRTA independent of hearing status and offer suitable intervention to treat dRTA as well as diminish the influence of SNHL on the child’s learning and communication in daily life.
Introduction
Renal Tubular Acidosis (RTA) is a pathological state that is characterized by chronic acidosis owing to the excessive loss of bases from blood due to impaired reabsorption of bicarbonates or built up of acids in blood due to impaired excretion of hydrogen ions [1]. It leads to malfunctioning of various cells due to systemic electrolyte imbalance, in particular imbalance of chlorine, potassium, and calcium. There are two types of RTA, i.e., proximal RTA (pRTA) and distal RTA (dRTA) that are associated with proximal and distal renal tubules, respectively [2].
The impairment in distal acidification process could be due to the secretory defects (failure of distal H+ secretion; also known as primary, inherited, classic or rate-limiting defect) or non-secretory defects (secondary defects with intact secretory mechanism). Secretory defect distal RTA is characterized by K+ secretion instead of H+ via apical membrane H+ ATPase or H+/K+ ATPase or the AE1 (HCO3-/Cl- exchanger) [3]. Two important types of primary dRTA are persistent and transient dRTA. An example of persistent dRTA is dRTA with sensorineural deafness [4]. Non-secretory defects include gradient, buffer, and voltage defect distal RTA. In gradient/back leak defect distal RTA, H+ secretion is active; however, H+ are back-diffused immediately after secretion. As compensation, loss of K+ takes place. It could be due to the abnormal membrane permeability. Buffer defect /low buffer type distal RTA also involves the loss of K+, since H+ secretion is impaired due to the increased luminal pH in the distal collecting tubule. Voltage defect distal RTA is characterized by hyperkalaemia due to the increased K+ reabsorption, and reduced H+ secretion [5,6].
Early-age dRTA is often inherited. It is commonly due to a defect in H+ ATPase or H+/K+ ATPase or the AE1 (HCO3-/Cl- exchanger) (), which are found to be associated with the following two mutation and phenotypic patterns, i.e. autosomal dominant and sporadic/recessive dRTA. In autosomal dominant dRTA, defect in AE1 is due to the mutation in SLC4A1 gene on Ch 17. The features of this dRTA include hypokalemia, hypercalcemia and nephrocalcinosis, hereditary spherocytosis (HS), South-east Asian ovalocytosis (SAO), short stature and osteomalacia [7,8]. Autosomal Recessive dRTA is due to following defects: (i) H+ ATPase B1 subunit defect (The gene responsible for this defect is ATP6B1 on Ch 2p13. These defects are accompanied with bilateral SNHL) [1]; (ii) H+ ATPase A4 subunit defect (The gene responsible for this defect is ATP6V0A4 on Ch7q33-34. These defects are accompanied with late onset of SNHL) [1]; and (iii) AE1 defect (The gene responsible for this defect is SLC4A1 on Ch 17. This defect is more prevalent in childhood and in Southeast Asians). Autosomal recessive distal RTA is accompanied with SNHL from early age [3].
Normal cochlear function depends on the homeostasis of cells and pH regulation [5]. The hearing impairment induced by the altered cellular homeostasis due to certain gene mutations has been reported [6]. These genes, in particular ATP6V0A4 gene, are involved in the production of certain protein complexes, such as vacuolar H+-ATPase (V-ATPase), which exist in the inner ear and nephrons. These complexes pump H+ across biomembranes and play a role in the regulation of cellular pH. Thus mutations in the ATP6V0A4 gene results in malfunctioning of the protein complex V-ATPase, eventually leading to pH disturbance in blood and cochlear fluid [7].
Results
CASE REPORT
A six year-old boy, born to a non-consanguineous Chinese couple, was admitted to our hospital with complaints of polydipsia and polyuria, of two years duration. His birth record showed that the boy was born through eutocia (G2P2) after a full-term gestation, having a weight of 4 Kg with no emergency treatment at the time of birth. Afterwards, his growth and development was normal. However at the time of admission, physical examination revealed growth retardation (height, weight <P3), and his visual acuity was within normal limit.
The supportive examinations showed alkaline urine, hypokalemia, elevated serum chloride, metabolic acidosis, and elevated alkaline phosphatase. Additionally, blood gas and electrolyte and close observation was regularly carried out. Double renal calcinosis could be identified in the B-mode ultrasound image. These findings revealed that the patient was suffering from renal tubular acidosis (I type). Urinary protein examination indicated moderate proteinuria, which is inconsistent with clinical manifestations of primary renal tubular acidosis (type I).
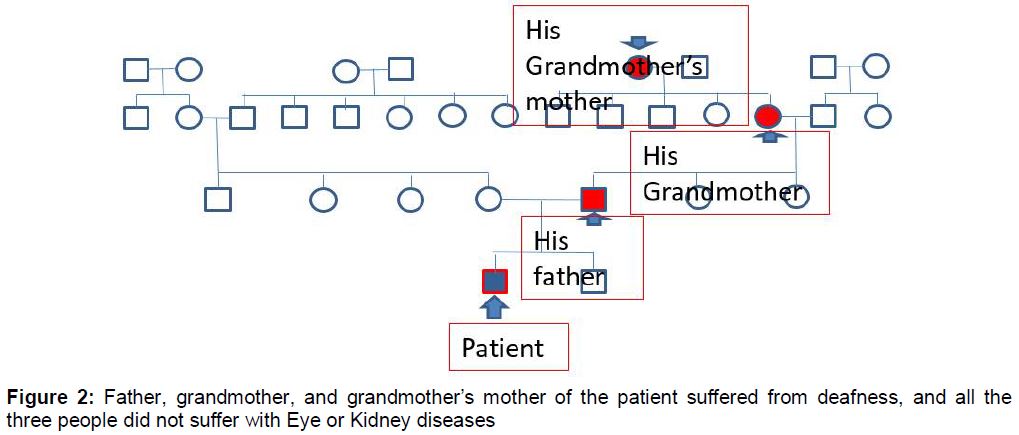
Combined with multiple renal cysts of both kidneys shown in B-mode ultrasound of the urinary tract, it was likely to be congenital kidney abnormalities, without excluding the possibility of genetic renal tubular lesions, which required complete genetic testing to confirm. Moreover, the enquiry of the patient’s family background revealed that his father, grandmother, and grandmother’s mother of the patient suffered from deafness, and all the three people did not suffer with eye or kidney diseases (). Genetic examination of renal tubule was scheduled to confirm the diagnosis it revealed.
Initially, informed consent was taken from the patient’s parents. Then DNA was extracted from the peripheral blood of the patient. Gene sequencing revealed 2 heterozygous mutations, c.1376C>T and c.1029+5G>A in gene ATP6V0A4. c.1376C > T (p.Ser459Phe) is a kind of missense mutation in gene ATP6V0A4. This mutation causes the change in amino acid number 459 in the protein coded by gene ATP6V0A4, from serine to phenylalanine. This mutation is extremely rare, and it has not been found in genomic sequencing databases for normal population. c.1029+5G>A is a kind of intragenic mutation near the cutting area of gene ATP6V0A4. This mutation is also extremely rare, and its frequency was 0.0023, 0.0023, and 0.0028. Distal renal tubular acidosis, related to gene ATP6V0A4, is a kind of autosomal recessive inheritance, and homozygous or compound heterozygous mutations could cause this disease. So the c.1376C>T and c.1029+5G>A mutations in gene ATP6V0A4 may cause the compound heterozygote, and it may be related to the reason for the disease, and further judgment needs more detailed clinical information. It was further suggested that Sanger sequence verification about these c.1376C>T and c.1029+5G>A mutations in Proband and his parents’ samples should be conducted to confirm whether these 2 mutations lie on different alleles, which form the compound heterozygous relationship. Moreover, renal assessment should also be advised, since this mutation is suspected because of the DRTA.
Discussion
The description and confirmation of RTA as a disorder were reported in 1935 and 1946, respectively, followed by the assigning of this term to the disease in 1951 [1-8]. It has been established that RTA should be an acquired or inherited disorder. The acquired RTA is induced by certain drugs, such as anticonvulsants, e.g valproic acid, anti-retroviral drugs, and anticancer drugs. Hence, close monitoring through periodic assessments of renal function and hearing of patients taking these drugs is compulsory [1]. Moreover, the inherited variety of RTA occurs through an autosomal recessive or autosomal dominant gene. Thus, the genetic testing and counseling are important attempts for offering the suitable intervention to treat RTA as well as suppress its effect on the patient’s daily life [2]. However, a high index of suspicion is required for the diagnosis of DRTA especially in the absence of hearing impairment.
There is an association between RTA and hearing impairment. The diagnostic results of RTA can be an indicator of impaired hearing, which progresses with time in RTA cases. Thus the audiological assessment of RTA patients should be undertaken at planned intervals [3]. A cohort study has reported the case of a person having ATP6V0A4 mutations showing the commencement of hearing loss in adulthood rather than during childhood [4].
Another study has reported that autosomal recessive dRTA develops in individuals with ATP6V0A4 mutations without emergence of hearing impairment [5]. The case study of another patient with normal hearing has been reported showing hyperchloremia, hypokalemia, nephrocalcinosis, and high urine pH in initial clinical diagnosis. Later on, genetic diagnosis revealed the ATP6V0A4 mutations that caused autosomal recessive dRTA [2].
Conclusion
ATP6V0A4 gene mutation study substantiated the autosomal recessive dRTA without hearing impairment in our patient. This case report highlights the importance of the significance of early diagnosis and genetic screening of recessive forms of dRTA independent of hearing status and offer suitable intervention to treat dRTA as well as reduce the effect of deafness on the child’s ability to exchange information in daily life.
References
Archives


News Updates